The Construction Operation and Accreditation of in vitro Science laboratory on Cosmetics
基于体外实验系统的安全和功效评价逐渐成为化妆品研发和检测的日常项目。体外实验室有其特殊的建设和运行要求,建设符合质量管理规范和认可准则要求的实验室,保证试验数据的可靠性和稳定性,可提高实验结果的公信力,促进整个行业水平的提升。上篇从体外科学在化妆品研发中的地位、实验室基本要求和认可制度、实验室硬件建设与生物安全、实验系统的稳定性与维护等方面介绍了基本要求和合理做法。
一、体外科学在化妆品研发中的地位
1、化妆品体外科学是大势所趋
经济全球化推动各国产品监管法规不断协调和融合,化妆品监管法规也符合这一趋势。众所周知的原因,欧盟于2013年率先实施了禁止化妆品动物测试的双禁令,即全面禁止动物测试,也不允许经过动物测试的化妆品在欧洲销售[1]。之后,全球先后超过40个经济体实施了全面禁令或有限禁令,既包括欧洲、东盟、澳新、日韩等发达经济体,也包括巴西、印度等新兴经济体,美国联邦部分州也推行了这一政策。目前,实施这一政策的国家和地区其化妆品市场已占到全球的60%以上。化妆品的研发和评价必然要淘汰旧有的动物模式,转向更快捷先进的体外替代技术。随着全球化的深度发展,以及“一带一路”倡议的推进,越来越多的化妆品需要进出国门,国内外产品同台竞争。统一测试方法,实施体外科学已迫在眉睫。
2、体外科学是化妆品研发的核心组件
摒弃传统的动物模式,转向采用新型的实验系统和方法。必然要吸收借鉴30年来现代生物医学技术的发展成果。正如2007年美国国家科学院“21世纪毒性测试:愿景和策略”中所指:毒性测试应转向基于人的或人来源细胞建立的新策略。如果以1991年欧盟成立专门验证机构动物试验替代方法验证中心(EURLECVAM)作为官方正式接受替代方法纳入法规监管开始,已累计投入替代方法研发经费超过60亿欧元。截至2020年3月,被世界经济合作和发展组织(OECD)采纳列入毒性测试指南的替代方法已超过35项,涉及11个毒理学终点。还有近70多项测试方法被国家、部门、协会或企业等不同层面接受[2]。如果加上个性化的功效性方法,体外科学占据了化妆品测试评价的主流。仔细研究这些方法的开发者不难发现其背后老牌国际公司的身影,如欧莱雅、宝洁、联合利华、巴斯夫、奇华顿、汉高、资生堂等。早在30多年前,体外生物技术强大驱动力已经植入了这些公司化妆品研发的基因中,使其能够从现代科技的发展中持续获益,同时收获与其国际地位相匹配的技术应用能力,科技软实力稳居第一梯队。本土企业在这方面刚刚起步,整个行业尚未形成共识,科技实力差距甚远。
3、体外实验室应具备的基本要求
法规接受的体外方法清单在不断扩大,加上全球体外试验市场以每年18%以上的速度增长,给体外新方法的开发和数据的互认提出了新的要求。如今,国内化妆品检测和研发使用体外方法基本上是拿来主义,已经非常方便。表面上看,使用者节省了从方法开发、验证、标准建立和法规认可的漫长的过程。但实际上,后来者缺少了对新技术投入的预判断,丧失了科研训练和人才队伍锻炼的机会,没有夯实可持续发展的基础。面对新技术怀疑观望、犹豫不定、盲目自信或患得患失是国内企业普遍的现象,正是缺少训练的表现。
从转化毒理学的发展史来看,目前监管机构接受的体外测试方法的科学性和可靠性是毋庸置疑的[3]。这些方法被不同监管机构所认可,如世界经济合作的发展组织(OECD)、国际标准化组织(ISO)、欧盟(EU)、中国国家标准(GB)和行业标准(化妆品安全技术规范和SN标准),而成为指南或标准。使得原来只有体内方法的标准体系加入了体外方法,并且比例还在不断增长中。因此,体外测试与体内测试对于数据的要求是一致的,即必须符合高质量、可重复和国际通用的要求。体外方法作为实验室检验检测体系的一部分,与其它试验方法一样需要遵循相似的实验室质量控制要求。实验室应具备基本的能力和资质,无论该实验室是第一方(企业内部)、第二方(官方机构)、还是第三方(社会独立机构)。
4、体外实验室的认可制度
国内外对于实验室能力的确认基本分为两类,一类是ISO体系,一类是GLP体系。ISO体系执行的最重要的标准是ISO17025,即《检测和校准实验室能力的通用要求》。在我国,唯一合法的认可机构是中国合格评定国家认可委员会(CNAS),依照《中华人民共和国认证认可条例》开展认可服务,根据认可准则要求开展检测校准实验室的认可工作。CNAS是国际认可论坛(IAF)、国际实验室认可合作组织(ILAC)和亚太认可合作组织(APAC)的成员。取得CNAS资格的实验室自然也获得了实验结果国际互认的通行证[4]。OECD的GLP体系文件很早就提出了GLP原则在体外研究中的应用指南。OECD指南的重点在于指导化学品的法规性安全测试,通过描述国际协调一致的实验方法,使之用于工业界、管理部门和第三方检测时都能获得明确和特征的化学品潜在危害信息[5]。
上述两种认可制度几乎覆盖了社会检测的各个方面,与每个人的生活息息相关。但是对于基于细胞、组织工程器官、干细胞和基因组的新兴技术,现有的认可制度存在不清明、不准确和非独立性的问题。鉴于此,2005年由ECVAM工作组起草发布了良好体外细胞培养规范(GCCP)文件[6]。2016年,EURLECVAM牵头启动了良好体外方法规范(goodinvitromethod,GIVIMP)的制订工作,历时3年,经广泛同行和社会意见,OECD于2018年12月正式发布了指南文本-No.286[7],成为体外方法研发、验证遵循的原则。因此,尽快建立符合中国国情的体外实验室认可制度非常重要。
认可的本质是向社会提供公正的服务,所以化妆品企业开展研发和检测工作,应具备或积极向认可的要求靠拢。获得认可的实验室(主要是提供校准、检验和测试服务的实验室)意味着:向社会各界证明其体系和技术能力能满足用户需要;可促进实验室提高内部管理水平、技术能力、服务质量和服务水平,增强竞争能力;使其能公正、科学和准确地为社会提供高质量的服务;减少和消除委托方(第二方)对实验室进行的重复评审或认可;通过国与国之间的实验室认可机构签订相互承认协议(双边或多边互认)来达到对认可的实验室出具证书或报告的相互承认,以此减少重复检验,消除贸易技术壁垒,促进国际贸易。
二、体外实验室的资质要求
1、独立地位和组织结构
外科学实验室或其所属的机构应有明确的法律地位。实验室应为独立法人或法人授权的独立组织中的一部分。注册和主要活动范围在国内的实验室应当获得权威机构的资质认定或(和)合格评定认可,检验检测机构应符合CNAS/GB/T15481-2000和ISO/IEC17025的认可准则的要求。实验室内部应有独立的质量管理手册,并在授权的范围内开展检测和校准活动。此外,还应当具有足够的能力保证从事体外研究和检测所需的场地、设备、环境和人力资源。应明确体外实验室在机构中的地位和关系,以及实验室内报告和信息的传递流向链。明确最高管理者在机构中的位置,明确在实验室中每个人的主要工作岗位和职责。明确质量负责人和技术负责人等关键人员的地位,并绘制出详细的内外组织架构图。
2、实验室建设的硬件要求
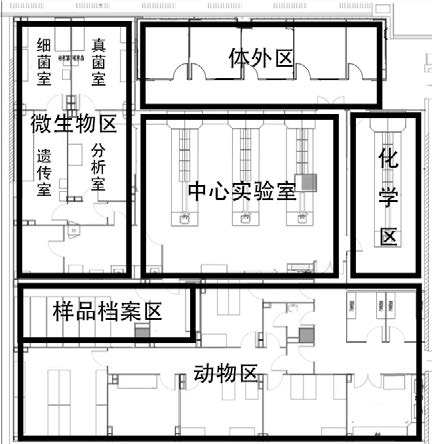
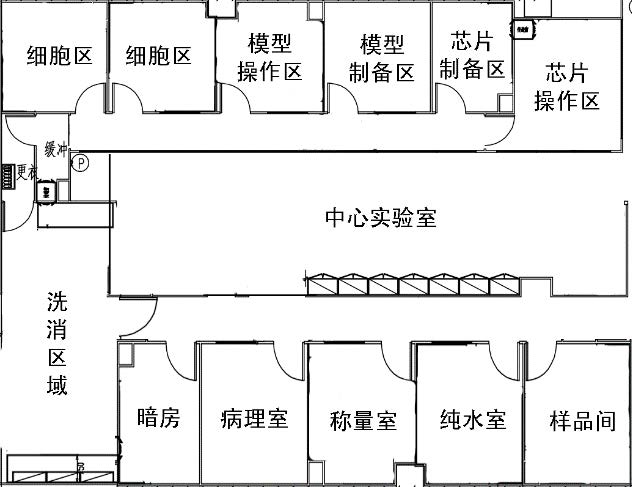
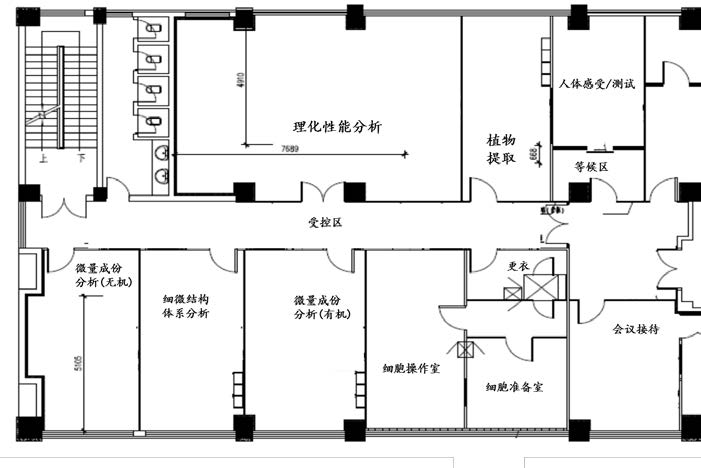
通常从事体外实验的机构还从事动物实验、化学检测和微生物检验等活动,应理顺与其它活动空间关系。图1是华代生物I期综合型检测中心实验室的总整体框架,整个实验区呈回字型布局。动物实验区,细胞实验区和化学分析分散在中心实验室周边,动物实验区与细胞实验区和微生物区通过走廊、样品室和化学分析区隔开一定距离,避免生物因素污染,但又相隔不远,方便必要时从动物获取实验材料。四个功能区域各自独立空调与通排风系统,避免交叉污染。如果实验室只从事体外科学相关研发和实验,应根据涉及的检测活动的范围确定实验室规模和布局,图2为华代公司的II期组织工程与器官芯片实验室的布局,设计时考虑到体外科学的专属性和前瞻性,设置了独立的细胞培养室,可从事原代细胞处理和基因操作。同时为了生产细胞、皮肤和芯片产品的需要,采用了微生物控制的净化装修。既满足检测需要,又兼顾和预留了科研与前沿技术的需要。此外,设施对于检测活动必要配套的样品室、数据室、图像分析室、制水室等也应按照要求合理布局。对于企业内部的实验室,除了质检品控外,还承担研发功能,微生物、理化和体外实验都是必不可少的,图3是广东三好科技有限公司研发中心布局。采用单走廊,分区和职能比较明显,细胞区设计紧凑,适合2-3人工作,满足企业体外实验室的基本要求。
图1 综合型检测中心平面图
图2 研发型体外实验室平面图
图3 企业体外实验室平面图
总之,设施必须适合于工作目的,它的大小、构造和位置都应该是恰当的,还应考虑建筑所处的城市大环境,包括风向、光照、楼层,以及周边是否有干扰实验室活动的潜在风险因素,如附近居民区、变电站、垃圾处理站等。还要考虑实验室供应链的便捷及与应急相关的处置措施的便利性。而国内企业比较在意的宣传、观察和展示功能应放在次要的位置。一个设计良好的设施,能有效确保在安全和有效的方式下产出高质量的实验结果。
设施投入运营之后,日常维护非常重要。应定期监测和维护实验室的各种环境因子,使之在相对稳定的状态下平稳运行,以减少环境波动对检测活动的干扰。如定期检查和更换初、高效过滤器;关键区域进行24小时温湿度监控并记录,洁净区的微生物要定期检测,以维持空调空气过滤系统的有效性;对有压力、光照和噪声有特殊要求的区域应做好实时或定期的监测。
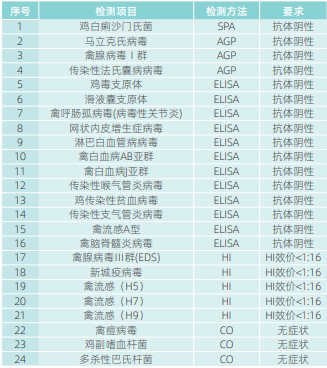
化妆品体外检测通常会用到生物材料,如现在比较普及的鸡胚、牛眼、细胞和人工皮肤等。对于细胞系或组织工程皮肤,应确保其来源清楚,确保没有微生物污染。需要长期保存的细胞株,应先进行支原体鉴定,再进行传代保存。对一次性使用的组织工程材料,如人工皮肤产品应做好批次检查并存档产品生物安全单。体外实验室还会接收或分离来自人或动物的细胞,如人的皮肤组织、动物的毛发、离体的牛眼或兔子的眼球或皮肤。应登记供体信息来源,并确保真实。对关键生物材料,如来自实验动物的材料,如鸡胚、兔皮肤或眼球、猪皮肤或眼球、小鼠毛囊,应注意这些动物可能携带的微生物会干扰实验结果,甚至可能具有人畜共患病的生物安全风险,如禽流感和大鼠出血热都是严重可能致死的传染病。应向供应商索取实验动物生产许可证,对鸡胚应附有检疫证明,符合有关标准,表1列出了国家标准(GB/T17999.1)要求的鸡胚微生物监测要求。实验室切记不可贪图便宜,从农户购买实验用材料,不仅实验结果没有保障,还有可能带来生物安全风险。对于畜牧业的废弃材料,如离体牛眼,应是合法和固定来源的,禁止向个人购买实验用生物材料。实验结束后生物材料应冷冻暂存,集中移交由合法的卫生处理场统一处理。
表1 用于实验的SPF级鸡胚微生物监测要求
备注:AGP-琼脂扩散试验;HI-血凝抑制试验;SPA-血清平板凝集试验;ELISA酶联免疫吸附试验;CO-临床观察
三、体外实验系统的稳定
体外实验系统不同于动物实验,有其独特的构成要素和要求。实验系统的稳定性是追溯和保证检测活动良好运行的开端。体外实验系统复杂多样,如原代细胞和细胞系、活的个体(鸡胚、鱼胚胎)、离体组织(牛角膜、动物皮肤)、重建模型(重建角膜和皮肤)、细胞组份(线粒体、细胞裂解液)和基因产物(如DNA提取物、PCR扩增产物)等,实验系统还包括用于支持生物系统生长、储存和处置的材料,如培养、储存材料和培养液。可见,体外实验涉及的生物学和非生物学材料非常多元,其操作复杂性、生物学安全等级及配套用品相差很大,这就决定了实验室既要制订规范化的程序,又要兼顾个性化的实验需求。归纳如下:
1、供应商评价
应向具有资质和信誉良好的供应商采购实验用品。生物材料通常来自权威的保藏机构,首选ATCC,国内的中检院和细胞保藏中心也可作为备选来源。易耗品的供应商应相对固定,包括培养材料(如培养皿,培养瓶),标准品、参考物质和阳性对照是实验室建立能力的重要保障,应保证这些试剂或材料来源可追溯,以及符合实验要求的等级。用于识别细胞身份、基准对照、诱导剂、监测检查试剂、重要添加剂(如生长因子、牛脑垂体提取物)等对实验运行或质量有重要影响的“关键试剂”,同样应建立供应商评价机制,确保有备选的来源。化学试剂,尤其是参考化学品应附有安全数据表(MSDS),列明重要的有关运输、接收和储存过程中的包装及处理的注意事项。对于有批次差异的试剂,如血清产品、生长激素、动物组织提取物,应在使用前进行质量控制,可采用室内细胞生物学效应、批次实验、化学检测等方法进行使用前检验。建立体外实验的关键试剂和耗材清单,并建立与之对应的合格供应商列表,可以有效控制体外方法中使用的消耗品和试剂的质量及稳定性。
供应商的资质、能力水平、量值溯源等是进行供应商评估的要点,评估合格后列入合格供应商列表。应定期对供应商进行再评价,主要评价其社会知名度、资质、质量、技术支持、服务效率、服务项目、价格、现场服务等方面。以确保合格供应商评价处于受控的、动态的和最佳的状态。
2、生物材料的控制要点
近年来,用于体外实验的系统发生了很大的变化,除了常用的细胞系,还包括人源化细胞、干细胞、重编程细胞等。而且,体外重建人体器官的使用也越来越广泛,如人工皮肤、类器官、干细胞产品和器官芯片产品。这些活性生物材料除了应关注生物安全风险之外,还应对其纯洁性和真实性进行鉴定,对关键生物材料应建立一对一的验收和管理标准。
1)纯洁性。生物材料应确保其纯洁,即无微生物污染。新引进细胞应进行检疫并确认支原体为阴性,还要证明没有细菌、酵母和真菌的污染。病毒可能来自原始供体、供应链,也可能由于使用者使用了污染的试剂或操作不当所致。一旦发现有微生物污染,应果断丢弃培养物,并彻底评估造成的风险程度,可适度付出一些代价,扩大清理的范围。
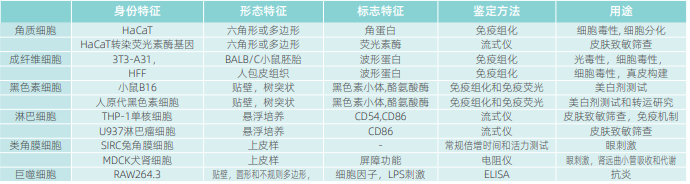
2)真实性。生物材料应具备代表其身份的特征,真实性还包括因细胞混合或基因组变异导致的不稳定,即遗传不一致和不稳定。因此建立早期细胞的DNA序列图谱(或保留原始组织样本)可为细胞识别和未来细胞变异追溯提供参考。基因的不稳定性在细胞培养中是绝对存在的,并且明智的做法应该是最小化维持细胞的传代次数(通常为p15-20),规定限制细胞的最大传代次数,并定期监测细胞的特征性能。在到达限制的传代次数时,应该从细胞库中寻找较低代次的细胞重新开始培养。在使用较高传代次数的细胞时,必须证明其完整性和功能性。除非有证据能证明细胞系功能正常,否则不明确传代次数的细胞是不允许使用的。有些实验会对细胞的基因组造成不稳定干扰,如皮肤衰老模型的实验通常采用电离辐射、癌基因诱导或过量UV照射诱导;或者采用转染的方法改造基因组获得工具细胞,如目前用于皮肤致敏实验的KeratinoSens和Lusen细胞,可能需要进行如单核苷酸多态性测序(aSNP)或比较基因组杂交(aCGH)等核苷酸或分子分析技术来核查其真实性。表2列举了常用细胞的真实性鉴定信息[7-9]。
干细胞的特殊性在于不可避免的含有二倍体和非整倍体细胞的混合物,采用基因测试可用于筛选干细胞在不同阶段间的变化,以及培养条件对干细胞分化产生的影响。在采用人类iPSC系构建类器官时,应监测是否存在重组基因的异位表达以及由非整合载体产生的用于消除载体的异位表达。尤其是研发型实验室应该特别关注这一点。3)功能性。实验系统应能在测试中呈现其应有的功能,并对毒性或功效性物质具备应答能力。如用于皮肤致敏筛查的THP-1细胞表面应具有在致敏原作用下产生CD54和CD86特征表达的能力,实验前应进行核查。如常用于抗炎试验的RAW264.3巨噬细胞在LPS的刺激下能产生炎性因子,如细胞无此功能则应废弃。
3、生物材料的验收
体外实验系统来源广泛,具有潜在的生物安全风险。实验室应对生物材料建立验收制度。来源于实验动物的组织或器官最好附有来源实验动物的质量合格证明,生产商还应提供当地科技主管部门颁发的生产许可证。进口生物材料应是合法渠道,附有海关许可文件、采购合同,并完整保留与商品相关的卫生和生物安全风险资料。图4是广州某公司某次从MatTek公司位于斯洛伐克的工厂进口皮肤模型的海关文件、模型质保负责人员签名和产品MSDS清单等文件。
图4 海关进口皮肤模型的证明
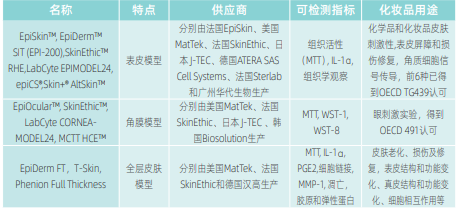
以皮肤模型的质控要求为例。皮肤模型的验收包括一般验收和技术验收。实验室在收到材料后立即验收皮肤模型的外包装、内包装、数量、状态(颜色、温度等)、所附材料(MSDS,生物安全证明)等。技术验收包括观察模型正反面是否出现破损和空泡等异常情况,并观察模型表面有无异常颜色或其他物质。模型孵育于培养液后,观察培养液是否渗漏至上层,合格的模型培养液不会漏至模型表面。皮肤模型应达到其声称的质量要求,不同商品化的模型特点不尽相同[2],如表3所示。质控要求也有差异,如同样是列入OECD指南492认可的角膜模型,采用了不同的质控方法[12]。表4所示,EpiOcularTM和MCTTHCETM模型采用了ET50的质控方法,而SkinEthicTM模型(中国有产生)和LabCyteCORNEAMODEL24模型采用了IC50的质控方法。图5为国产EPISKIN模型随商品的批次质控表。皮肤模型应满足多层角质结构,活性保持在>60%,对刺激剂反应一致(应向供应商索要上述证明)。出于公正和受监督的原则,皮肤模型生产者不应从事使用该模型的外部检测工作。
表2 化妆品实验常用细胞的真实性鉴定
表3 可用于化妆品安全功效评价的商品化重建皮肤和角膜
图5 皮肤模型质控要求是验收的重要依据
4、实验材料日常管理
实验室应建立维护实验系统稳定性的质量文件,最好有相对独立的良好细胞培养规范[10],并强制执行,列入规范的内容包括培养基测试、血清的质控;新引入细胞或组织的接收、测试和接收标准;试验系统污染的处置;试验系统的变动记录;收藏、存贮和处置研究废弃物的设施和设备;培养用具清洁去污和处置规程等。
在常规处理和维护过程中,应记录细胞系统的生长、生存特征(如细胞活力、倍增时间等)和传代培养细节(例如传代日期、传代时间间隔、播种密度、传代次数等)并在研究报告中记录,因为它们是结果完整可追溯性所必需的。测试系统供应商提供的文件应与历史数据一并提供,并用于确定验收标准。
每个实验设备应制定SOP,包括如何解冻,处理,计数,维护和保存细胞系的细节。例如,应建立程序明确传代次数以及判断细胞储存活力的测试方法。
原代细胞和组织材料的获取必须合法并符合伦理要求,包括人的和动物的伦理要求。对于具体实验还应考虑供体的差异,并且每批次都应确认质量或控制关键功能。应特别注意是否有微生物污染,如实做好记录。作为生物组织质量控制的重要部分,它们的分化状态也应记录,包括采用的测定方法及培养过程中观察到的详细信息,如形态学、组织化学、细胞标志物、特异性组织功能和细胞-细胞基质相互作用等。体外实验系统的规范性管理是体外实验室可以持续稳定运行的前提。
致谢:感谢国家认监委科技支撑项目《体外方法实验室认可评价关键技术的研究》(2017RJWKJ10)和广州市经济技术开发区国际合作项目(2017GH11)的支持。
参考文献 :
[1]程树军,焦红.实验动物替代方法原理与应用.北京:科学出版社,2010
[2]程树军.化妆品评价替代方法标准实施指南.北京:中国标准出版 社,2017.
[3]Curren RD,Eskes C,Cheng S, et al. Contributions to the Development of Alternatives in Toxicology in China and Brazil, in The history of alternative test mehods in toxicology,Acaemic Press,2018
[4]史光华,吕京,程树军.关于实验动物机构认可工作的思考.实验动物与比 较医学,2012,32 (1):73-77
[5]程树军,焦红,潘芳.GLP原则在体外毒理学评价实验室的应用.中国比较 医学杂志,2008,18(11):58-61
[6]Eskes C, Boström AC, Bowe G, et al. Good cell culture practices and in vitro toxicology. Toxicol. In Vitro,2017, 45, 272–277.
[7] OECD.2018. Guidance Document on Good In Vitro method practices, Series on testing and assessment, No.286,OECD Publishing,Paris.
[8]Hirsch C, Schildknecht S.In Vitro Research Reproducibility: Keeping Up High Standards.Frontiers in Pharmacology,2019,10:1484.
[9]Gutbier, S., May, P., Berthelot, S., Krishna, A., Trefzer, T., Behbehani, M., et al. (2018). Major changes of cell function and toxicant sensitivity in cultured cells undergoing mild, quasi-natural genetic drift. Arch. Toxicol. 92(12), 3487–3503.
[10]Pamies D, Bal-Price A, Chesné C, et al. Advanced good cell culture practice for human primary, stem cell-derived and organoid models as well as microphysiological systems. ALTEX,2018, 35 (3), 353–378.
[11]OECD. 2019. Test Guideline No. 492:Reconstructed human cornea-like epithelium (RhCE) test method for identifying chemicals not requiring classification and labelling for eye irritation or serious eye damage. OECD Publishing
程树军 马莎莎 陈志杰
1. 上海交通大学医学院
2. 上海市毒理学会
 供需大厅
供需大厅
 登录/注册
登录/注册 供应商登录
供应商登录