 供需大厅
供需大厅
 登录/注册
登录/注册 供应商登录
供应商登录











荣格工业资源APP
了解工业圈,从荣格工业资源APP开始。
欢迎来到荣格工业资源网!

供需大厅

登录/注册

公众号

更多资讯,关注微信公众号

小秘书

更多资讯,关注荣格小秘书

邮箱
您可以联系我们 info@ringiertrade.com

电话
您可以拨打热线
+86-21 6289-5533 x 269
建议或意见
+86-20 2885 5256

顶部

荣格工业资源APP
了解工业圈,从荣格工业资源APP开始。


细胞和基因治疗可以为患者提供“治本”的疗法,一次性解决疾病根源,而不是让患者接受长期的“治标”治疗,从而大大改善了患者的生活。许多细胞和基因治疗产品是针对罕见病的,因此细胞和基因治疗产品很多申请了加速审批。
这些因素让细胞和基因治疗行业注入勃勃生机,并推动行业深入了解不断变化的法规要求,以便加快产品的上市进程。
然而,要在不牺牲质量的前提下实现这些产品的商业化,行业和监管机构之间需要进行良好的合作和沟通……
Part 1
病毒载体产品的特性
病毒载体是经过改造的病毒。病毒载体可以作为将遗传物质传递到细胞中的工具,在基因和细胞治疗中,用于治疗特定疾病。
病毒载体需要保证完整,遗传物质才能够被运送到特定的组织中,进入靶细胞。
腺相关病毒(AAV)基因组早在上世纪 80 年代就被分离和克隆。上世纪 90 年代,新型工程化 AAV 衣壳的重大进展和大规模生产的实现以及纯化效率的提高,使得基因治疗药物成为可能。其中,一款靶向肝脏的血友病 B 基因治疗药物已经成功,该药物采用了 AAV 载体工艺,并通过静脉注射给药。

细胞和基因治疗产品的生产过程与蛋白质/单克隆抗体(mAb)的生产过程类似,但病毒载体的生产更为复杂。
例如,用于病毒载体工艺开发的实验室的设计必须达到生物安全二级(BSL2)标准。BSL-2 实验室废弃物的需要更加严格的消毒和管理。
此外,病毒载体生产十分依赖一次性技术。
一次性技术是一个封闭的系统可用于多种生产平台,并且在多产品共线生产时可以有助于制定整体污染控制策略。
确保一次性系统无菌对提供安全的药品至关重要。必须有相应的流程来确保所用的一次性生产设备已通过无菌验证,除了通过供应商审计对生产商进行资格认证外,还要评估无菌验证方案。
进行病毒载体生产的工厂还必须配备单通空气流的空调通风系统,专门用于隔离和防止交叉污染。
在使用一次性技术的过程中也会遇到许多难题。
例如:
许多一次性系统采购自单一供应商,部件难以于其他系统兼容;
如何建立适当的可提取物/可浸出物研究,从而确定一次性系统是否与生产的生物制剂相容;
如何在生产过程中保持一次性系统的封闭性从而防止微生物污染?
美国食品药品监督管理局(FDA)已经发布了《现场观察报告》(483 缺陷报告),指出生产商没有意识到部件不兼容导致一次性系统出现泄漏。
因此对于生产过程中或运输/搬运过程中部件使用不当而可能导致的泄漏,生产商需要格外小心。
最近,FDA 表示正考虑参考 510(k) 流程,允许申请人使用批准的生物制剂生产平台生产基因治疗药物,任何批准后的改动都要经过 FDA 的再次审查,确保生物制剂符合 FDA 的药效评价标准并且不会增加安全风险。此流程可以简化生产商产品开发流程,同时促进全球采用统一的方法。
事实上,PaVe-GT 项目是由美国国家转化科学促进中心 (NCATS) 牵头的平台载体基因治疗试点项目,旨在确定采用标准流程是否能显著提高基因治疗试验初创公司的效率。
目前,该项目将 AAV 用作治疗四种罕见病的标准化平台(采用相同的衣壳和生产方法)。目的是使用同一种病毒载体、生产工艺和团队,开发出节省时间和资源的生产平台。
单一生产平台的优势包括但不限于简化临床前研究工作、使用相同的生产工艺以及治疗多种不同的疾病。
单一生产平台的优势包括但不限于简化临床前研究工作、使用相同的生产工艺以及治疗多种不同的疾病。
在打造单一生产平台的同时,细胞和基因治疗产品的上市也面临着重重挑战,比如推动已获得快速通道认定的罕见病和超罕见病药物的开发。
快速发展的细胞和基因治疗行业的另一个特色是质量管理体系(QMS),该体系可根据市场对可扩展性和开发速度的具体需求进行定制,并且还保持了稳健性。与传统的商业化路径相比,细胞和基因治疗行业压缩了 II 期开发、III 期开发和工艺验证的时间。
可扩展 QMS 的一大特性就是包含一个流畅的技术转移流程,这一流程列示了实现工艺验证的关键阶段和计划,而并非只是进行简单的勾选练习,它有助于训练员工在可扩展的环境中的工作能力。在确保妥善记录和批准所有风险和相关缓解活动的同时,该体系能够适应不同的审批途径,这对产品的成功至关重要。
Part 2
改善先进疗法的临床环境并加强监管
截至 2019 年底,正在进行的再生医学/先进疗法的临床试验超过 1,000 项,在全球范围内,活跃在基因和细胞治疗以及其他再生医学领域的公司筹集了近 1,000 万美元。
FDA 也发现研究性新药 (IND) 申请正在增加。
FDA 前局长 Scott Gottlieb 预计,到 2025 年,每年将有 10 至 20 种细胞和基因治疗产品获准新药申请。
以上数据不仅表明了这些基因和细胞治疗产品的成功,同时也表明了 FDA 大力支持基因和细胞治疗产品的开发,因为随着行业快速发展,其规模和范围需要正式和非正式的指导。
FDA 预计的这一发展情况也与麻省理工学院的预测不谋而合——到2030 年,将有 40 至 60 款基因和细胞治疗产品上市,惠及超过 50 万名患者。
最近,FDA 最终确定并发布了几份细胞和基因治疗指导文件,向需要的公司提供更多信息。
这些文件包括:
2020 年发布的 FDA 最终指南
人类基因治疗研究性新药 (IND) 申请的化学、生产和控制 (CMC) 信息
在产品生产和患者随访期间,测试基于逆转录病毒载体的人类基因治疗产品能否复制逆转录病毒
视网膜疾病的人类基因治疗
罕见病的人类基因治疗
人类基因治疗产品的患者长期随访管理
血友病的人类基因治疗
除了这些指南外,FDA 还通过审查途径和专门的产品认定来加快申请审理进程,确保一旦确定这些病症的疗法疗效大于风险后,便可批准该疗法并使其惠及患者。
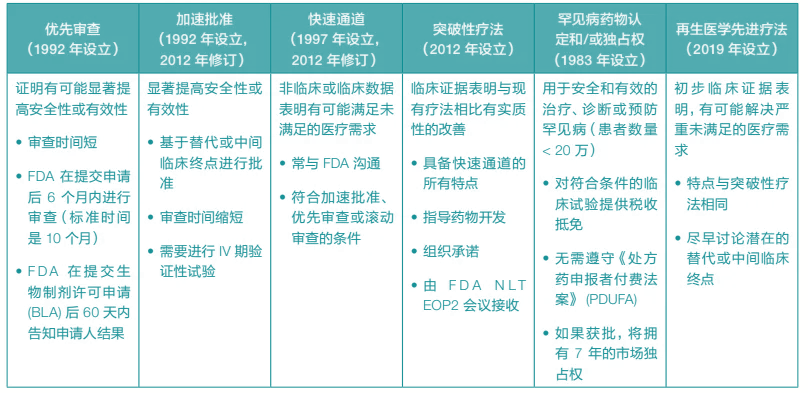
FDA还表示愿意与希望深入了解这些流程并达到所需里程碑的公司合作。
例如,生物制品评价和研究中心 (CBER) 已经启动了一项新的会议程序,名为“CBER 产品监管建议初步专项会议 (INTERACT)”,此会议可以让生物制药公司在早期产品开发时就与 CBER 沟通而不用等到 pre-IND 会议的时候。这在科技迅速发展且需要向患者快速给药的情况下可以让公司尽早获得 FDA 的指导。
Part 3
细胞和基因治疗面临的监管和 CMC 挑战
CBER 和药品审评和研究中心 (CDER) 坦言,他们过去一直难以跟上药物创新的步伐,难以在加快评估的同时保持质量和患者安全,也很难确定风险-效益评估的方式。
在最近的会议上,CDER 讨论了不完整的 CMC 文件包会在提交时影响生物制剂申请的审理,导致监管机构提出更多问题,甚至可能延长审理时间。
从行业角度来看,由于项目速度加快,要使 CMC 开发进程与临床开发保持同步,协调开发时间、供应链和实际限制条件,并提供含有关键 CQA 信息的完整产品表征数据,一直是一项充满挑战的工作。
然而,尽管存在这些挑战,监管机构与细胞和基因治疗行业之间加强沟通,只会有助于将这些令人惊叹的疗法更快地推向市场,而不会出现其他负面影响,我们必须共同努力实现这一目标。
作者:Monica M. Commerford 博士,赛默飞病毒载体服务法规事务部经理


